Summary
Replimune (NASDAQ: REPL) is a Massachusetts-based biotechnology company focused on developing oncolytic immunotherapy solutions through its proprietary RPx biological platform. The Food and Drug Administration (FDA) set a Prescription Drug User Fee Act (PDUFA) Action Date for Replimune's primary asset – RP1, or vusolimogene odeparepvec – for earlier this week; Novana Investment Management acted on a bullish conviction for RP1's regulatory approval through a bull call spread position, which ultimately expired out-of-the-money. The net loss on this trade was $1,595.00 (0.158% of our total assets under management), the difference between the dollar-cost average of the purchase of two sets of call options (ten contracts, totaling $2,197) and the dollar-cost average of the sale of two sets of call options (ten contracts, totaling $602).
About Replimune
Replimune's core value is derived from its in-house RPx platform, a biological platform powered by an engineered Herpes Simplex Virus type 1 (HSV1) backbone. The scientific thesis underlying RPx is that its ability to insert targeted transgenes has the potential to revolutionize general cancer treatments by encouraging patients' immune systems to destroy local tumor cells — alone, or in conjunction with complementary therapeutic systems. All three of Replimune's biologic assets, RP1, RP2, and RP3, are built off of RPx and are at various stages in the clinical pipeline for applications spanning advanced melanoma, non-melanoma skin cancers, skin cancer organ transplants, metastatic uveal melanoma, and hepatocellular carcinoma. The company has been operating for 10 years to-date, having raised an aggregate $442 million in capital across its Series A round, Series B round, initial public offering (IPO), private investment in public equity (PIPE) transaction, and public share/warrant offerings.
About RP1
RP1 (vusolimogene odeparepvec) is comprised of three distinct biological entities: an engineered HSV1 backbone, a Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) cytokine, and a modified glycoprotein from the Gibbon Ape Leukemia Virus (GALV-GP R-). Replimune has been developing RP1 for at least five years, as dated by their 2019 research article, "Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1," in the Journal for Immunotherapy for Cancer. Since then, Replimune has entered RP1 into three clinical trials – IGNYTE, IGNYTE-3, and ARTACUS – for treatments spanning advanced melanoma, non-melanoma skin cancers, and skin cancer organ transplants. In the case of approval for this PDUFA action, RP1 would be administered alongside Opdivo (nivolumab) for advanced melanoma patients who have previously exhausted anti-programmed death protein 1 (anti-PD-1) therapeutic options; Replimune would be a second mover in the space, competing directly with Iovance Biotherapeutics's AMTAGVI (lifileucel), a one-time tumor-infiltrating lymphocyte (TIL) therapy.
The Basis for Our Trade
RP1's leading clinical pathway is for the treatment of advanced melanoma, with its application-specific IGNYTE trial having concluded a supposedly successful Phase 3 testing (read our second piece, "How FDA Politics Upended Replimune's RP1," for more details about this). Furthermore, RP1 is Replimune's leading clinical asset, having progressed further in clinical stages than both RP2 and RP3. Therefore, the FDA's decision for RP1's commercialization would be a significant tell for investors as to how the rest of Replimune's HSV1-based therapeutic assets would fare with regards to regulatory approval. The FDA set July 22nd as its PDUFA Action Date for RP1, which was the event around which we built our trade. We believed that the FDA's decision, regardless of directionality, would significantly impact Replimune's share prices, given the company's 10-year cash burn and RP1's status as its first clinical asset to potentially commercialize.
Scientific Due Diligence
Melanoma is a variation of skin cancer, one that develops when melanocytes – pigment-producing skin cells – take on a cancerous form. Currently, it represents the fifth-most common form of cancer in the United States, with approximately 100,000 new cases and 8,000 deaths annually. Survival in early, localized cases (Stages 0 and Stage I) are high: survival cases hover around 90% for 5-to-10-year survivability. Rates begin to decline significantly after the melanoma expands, however. The 5-year survival rate for further-developed cases (Stage II, Stage III, and Stage III) dip to 45-79%, 24-70%, and 7-19%, respectively. Though early intervention usually ensures improved outcomes, patient responses to the leading primary treatment, anti-PD-1 therapy, are incredibly unreliable. Immune checkpoint inhibitors only yield successful responses for just over 50% of melanoma patients, and initial anti-PD-1 therapy administration increases the timeframe during which patients' conditions are prone to deteriorating. For those whose immune systems do not respond to anti-PD-1 therapy, secondary therapeutic methods such as alternative immunotherapies, targeted therapies, or chemotherapies must be used.
Among the most important metrics for oncolytic therapies are overall response rates (ORRs), complete response rates (CRRs), duration of response (DOR), and treatment-related adverse event (TRAE) grade distributions. Shown in the table below are these metrics for RP1 and its only competitor, AMTAGVI:
| Oncolytic Therapeutic | ORR | CRR | DOR | TRAE Grades I – II | TRAE Grades III – IV |
|---|---|---|---|---|---|
| AMTAGVI | 31.4% | 5.9% | 36.5 months | > 80.0% | 40.00 – 77.00% |
| RP1 | 33.6% | 15.0% | 21.6 months | 76.6% | 12.8% |
RP1 had stronger response rates amongst patients, as well as less severe adverse event grades and frequencies, than AMTAGVI. These clinical endpoints satisfied the FDA's stated criteria for a Breakthrough Therapy candidate, which is that "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s)." Additionally, we verified the validity of the results from Replimune's trials by scrutinizing their patient sizing: we found that 100 to 200 patients is the historical range that regulators have accepted for single-arm registrational oncology trials when the effect size is large and there is high unmet need; IGNYTE's Phase I/II trial enrolled 140 patients, which did not raise any suspicion.
Financial Due Diligence
We carried out the following back-of-envelope calculation to calculate Replimune's "fair" share price:
Price_today = P(approval) × Value_if_success + (1 – P) × Value_if_failure
To do this, we had to derive four intermediate values: the probability of RP1's approval, the probability of RP1's rejection, and Replimune's share price following FDA rejection. We employed a simple heuristic to approximate the probability of RP1's approval. First, we obtained a base probability by identifying the broad-based approval rate for biologics therapies in the same disease area and phase transition as RP1. Next, we identified tailwinds and headwinds to RP1's approval, adding and subtracting to the base probability accordingly.
According to data from "Clinical Development Success Rates and Contributing Factors 2011–2020" by BIO (Biotechnology Innovation Organization), Informa Pharma Intelligence, and QLS Advisors, the NDA/BLA (the latter of which RP1 had already attained) to Approval rate for Oncology is 92.0%, with a 324 sample size. Therefore, our base rate assumption for probability of approval (POA) for RP1 was 92.0%.
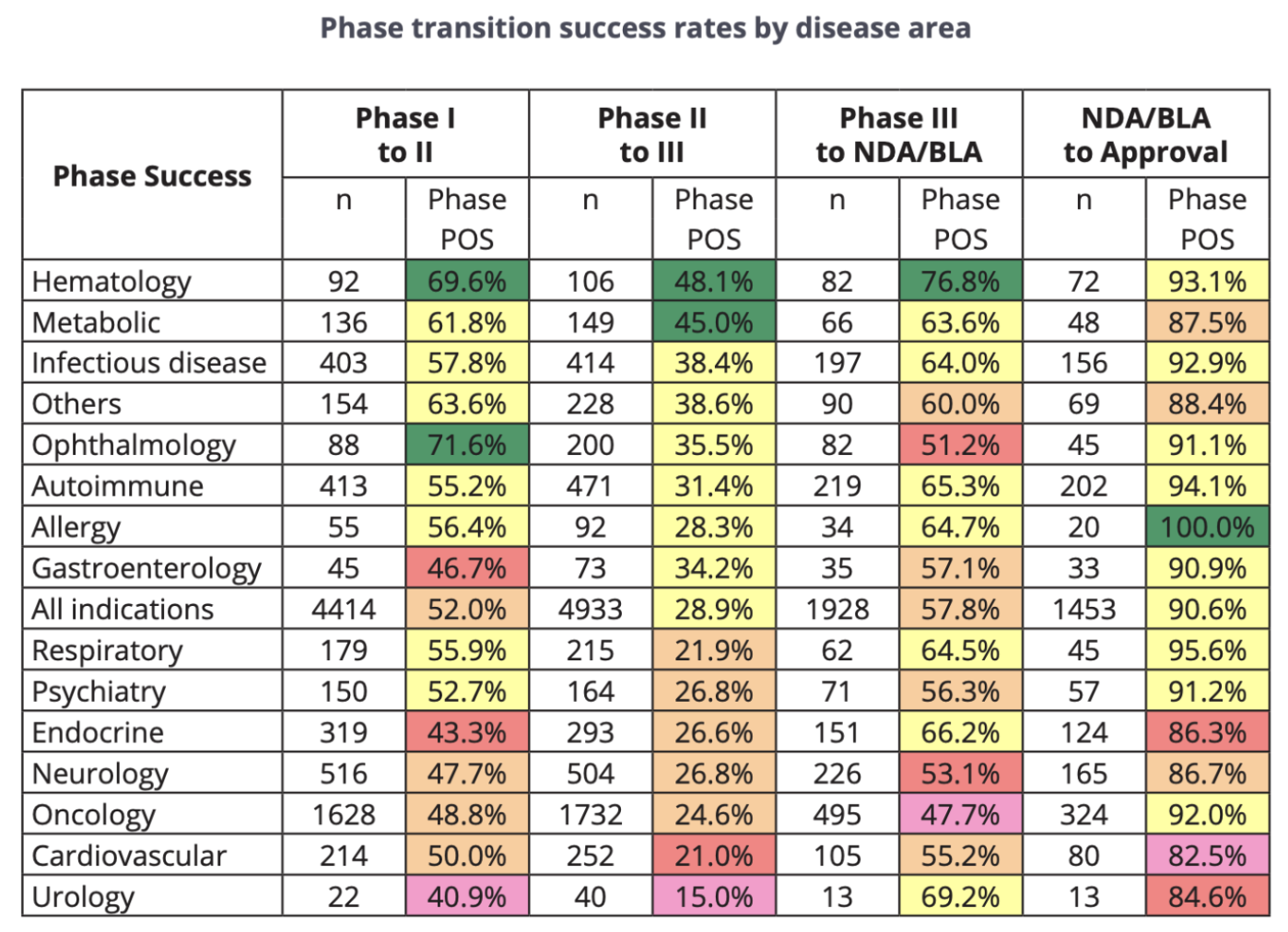
A series of designations granted to RP1's advanced melanoma BLA by the FDA positively skewed our base POA: General Accelerated Approval, Breakthrough Therapy Designation, and Priority Review all have historical associations with higher-than-average approval rates. According to a report from the American Association for Cancer Research, 63% of cancer drugs that had been granted general accelerated approval between 2013 and 2017 were eventually converted to regular approval. Similarly, the FDA reported that 54% of drugs that obtained breakthrough therapy designation were converted to regular approval. NDAs/BLAs with priority designation also seem to yield higher-than-average approval rates, though the approval rate differential between priority and standard NDAs/BLAs has tended to fluctuate over time (as shown in the graph below).
The headwinds that we identified were all CMC-related (Chemistry, Manufacturing, and Controls). During our diligence process, we were able to find examples of last-minute CMC concerns that created delays to therapeutic commercialization: the approval of Amgen's T-VEC (Imlygic), for example, was postponed three months "due to a request… for additional existing manufacturing data." In fact, the FDA's own public-facing guidance on gene-therapy CMC states that insufficient viral-vector CMC data may result in clinical holds or BLA review delays. We incorporated a non-zero probability that the FDA would find outstanding issues with Replimune's product CMC processes into our calculations, with missing data on the manufacturing process, insufficient quality controls, or incomplete testing results serving as a few possible situations that could realize this outcome.
Taking into account our 92% baseline POA, precedent-based tailwinds, and regulatory headwinds, we estimated RP1's probability of approval of 94-96%; we believed that the tangibility of RP1's General Accelerated Approval, Breakthrough Therapy Designation, and Priority Review designation benefits outweighed the significance of the outlier CMC-related fail cases that we found during our research. Accordingly, our estimation of RP1's probability of rejection was 4-6%.
We based our estimations of Replimune's share price upon RP1 approval solely off of the consensus 12-18 month analyst price target from Yahoo Finance— this value was $21.88. Calculating Replimune's share price upon RP1 failure was a more nuanced process, however. First, we calculated REPL's undiscounted cash per share: the company's latest reported cash and short-term investments totaled $483.8MM and fully-diluted shares (as of its FY-2025 10-K) totaled 80.6MM, yielding an undiscounted cash per share of ~$6.00. Historical precedent demonstrates that share prices trade at between 60-80% of cash-per-share following a Complete Response Letter (CRL) issuance, meaning REPL would likely trade at $3.60 to $4.80 in the case of a regulatory delay. Complete rejection, on the other hand, has a precedent of post-event share value of 30-50% of cash-per-share, which would discount REPL to $1.80 to $3.00. These previous calculations fail to account for the presence of secondary and tertiary pipeline assets, however. Another factor we incorporated into our calculations was our estimate of Replimune's $0MM to 100MM of pipeline value ex-RP1 via its RP2 and RP3 assets (dependent upon FDA decision), leading to the following results:
- Moderate Discount (0.75 × cash-per-share + 100M pipeline): (0.75 × 484) + 100 ≈ $463MM and $5.75 price-per-share.
- Harsh Discount (0.5 × cash-per-share + $50M pipeline): (0.50 × 484) + 50 ≈ $292 MM and $3.60 price-per-share.
- Complete Failure (0.3 × cash-per-share + $0M pipeline): 0.30 × 484 ≈ $145 MM and $1.81 price-per-share.
For our final estimate of Replimune's share price upon RP1 failure, we used the mean between our Moderate Discount and Harsh Discount scenarios as a proxy ($4.68 price-per-share).
Having derived all four intermediate values – the probability of RP1's approval, the probability of RP1's rejection, and Replimune's share price following FDA rejection – needed for our final fair value estimate, we were able to substitute accordingly into our back-of-envelope equation as follows:
0.95 × $21.88 + 0.05 × $4.68 = $21.02 fair value estimate.
Determining Our Position
Replimune's shares were trading at $12-13 per share in the days leading up to RP1's PDUFA action date — a significant discount to our internal estimates of Replimune's fair share price. Additionally, we noted that Replimune's most immediate options contracts had the most market activity at call and put strike prices at $12.50 and $7.50, respectively. Discounted live share price and a tighter-than-expected call-put spread led us to considering the following two options trading strategies: 1) a long straddle; and 2) a bull call spread.
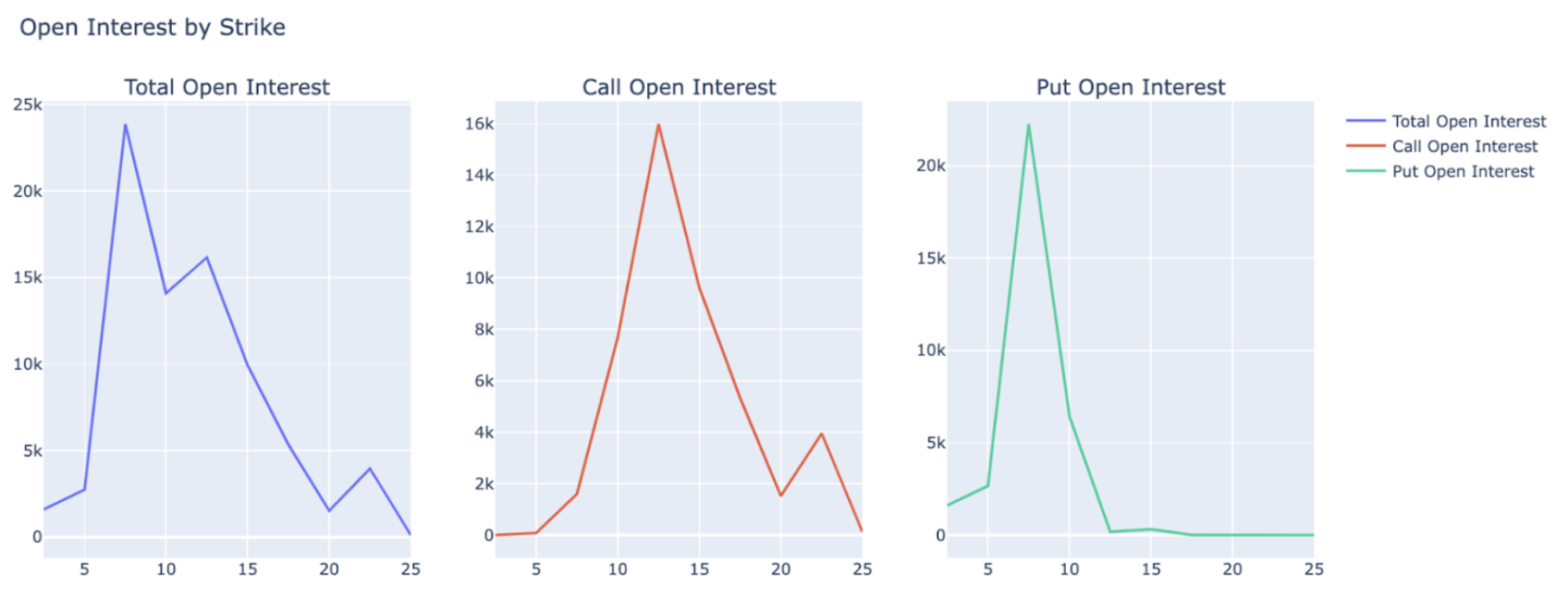
A long straddle would have consisted of the simultaneous purchase of REPL call options and put options with the same strike price and expiration date and benefitted off of volatility post-event greater than what was priced in. Given that the call and put strike prices with the greatest open interest were between our internal conditional price-per-share estimates ($4.68 in the case of delay/failure and $21.88 in the case of approval), this was a viable strategy. Alternatively, a bull call spread would have consisted of the simultaneous purchase and sale of REPL call options with varying strike prices and the same expiration date and would have benefitted from a large, directional price movement of the underlying asset.
The strategy that we executed was a bull call spread. We purchased ten $12.50-strike-price call option contracts with a $2.197 dollar-cost-average and sold ten $17.50-strike-price call option contracts with a $0.602 dollar-cost-average. The net cost – and maximum loss – of this trade was ($2.197 – $0.602) × 1000 = $1,595. Our maximum profit threshold was [($17.50 – $12.50) – ($2.197 – $0.602)] × 1000 = $3,405, yielding a reward-to-risk ratio of approximately 15:7 and a breakeven price of $14.10.
Trade Result
The FDA issued a CRL for RP1's BLA, which drove Replimune's day-over-day share price down 78%. Novana's bull call spread position expired out-of-the-money, leading to a net loss of $1,595.00 (0.158% of our total assets under management). We unpack our post-event analysis of Replimune's regulatory failure here.